
Разработка единой теории молекулярных спектральных и химических процессов
а) Развитие теории внутренних движений атомов в крупных молекулах и их ИК и КР спектров
Движения атомов в молекулах являются важнейшими в микромире, и ими определяется большинство молекулярных процессов, а на применении соответствующих спектров базируется целый ряд базовых методов исследования микромира. Расчёты колебаний и внутренних вращений в молекулах широко использовались и используются для решения громадного числа практически важных проблем. Более того, развитие общей теории колебательных спектров сложных молекул послужили основой для создания теоретической базы многих разделов теоретической физики микромира. В этой области науки после выхода второго издания книги «Колебания молекул» (1972 г.) был накоплен большой дополнительный материал о расчёте очень больших (без ограничений на число атомов) нерегулярных молекулярных структур, о колебаниях большой амплитуды, внутренних вращениях и др. Этот материал разбросан по многим статьям в разных журналах и его обобщение, монографическое изложение представляется, поэтому, крайне важным. Эта работа была выполнена в 2007 г.
Обобщены результаты и подведены итоги работ по созданию теории, вычислительных алгоритмов и удобных для реализации на ЭВМ методов вычислений характеристик внутренних движений атомов в крупных молекулах и их ИК и КР спектров. По результатам этой работы подготовлена монография «Колебания молекул» (изд. 3), которая содержит обширный материал, полученный одной научной школой и поэтому отличающийся единством и взаимосвязанностью. В книгу вошло большое число не только ранее выполненных исследований, но и новых, специально проведённых в процессе ее написания. По сравнению со 2-м изданием содержание монографии обновлено более чем на 70 %. Книга не является обзором области, а целиком базируется только на оригинальном материале, полученном научной школой за многие годы.
Монография состоит из 16 глав и заключения; ее содержание по главам следующее.
В первой главе рассматриваются базовые методологические положения теории молекулярных спектров. Особое внимание уделяется принципу дополнительности Н. Бора, понимание которого является необходимым для выбора оптимального пути построения теории и методов расчёта любых свойств молекулярных систем. В специальном параграфе обсуждаются взаимосвязь и дополнительность полуэмпирических и ab initio подходов.
Вторая глава посвящена общим вопросам теории внутренних движений в молекулах: построение уравнения Шредингера в системе криволинейных координат, адиабатическое приближение, гармоническое приближение, разделение видов движений и др.
В главе 3 описываются методы вычисления кинетической составляющей гамильтониана, вводятся внутренние (естественные) координаты разного типа: от координат растяжения связей до координат, включающих центры масс атомных групп. Даётся классификация колебаний. Приводится целый ряд важнейших для приложений соотношений декартовых и внутренних криволинейных координат.
В 4-й главе рассматриваются свойства матриц силовых постоянных и коэффициентов влияния, устанавливаются связи матриц силовых постоянных для разных систем координат, анализируется влияние неадиабатичности и др.
Главы 5 и 6 посвящены вопросам, связанным с симметрией молекул и её использованием для разделения переменных в колебательной задаче. Подробно излагается отличающийся большой наглядностью матричный метод построения координат симметрии, вводится понятие вектора симметрии и др.
Следующая глава содержит описание методов вычисления частот и форм колебаний и производных от них по параметрам при использовании как независимых, так и зависимых внутренних координат. Проводится пример решения задачи о колебаниях для объекта с очень большим числом атомов и числом зависимых координат.
В главе 8 рассматривается важная проблема об условиях появления характеристических по разным признакам (частоты, формы) колебаний и проблема расчёта колебаний нерегулярных структур любой сложности (молекулярные кристаллы и очень крупные молекулы) с использованием банка молекулярных фрагментов и теории возмущений в матричном и операторном вариантах. Показывается, что ряд теории возмущения Шредингера получается как частный случай при малых углах поворота метода вращения Якоби.
Главы 9-13 посвящены теории и методам расчёта интенсивностей полос и линий в ИК и КР спектрах. В первых параграфах рассматривается квантовая теория взаимодействия электромагнитного поля и вещества, причём показывается, что спектры КР могут трактоваться как результат взаимодействия молекулы с модулированным полем. Обсуждаются общие характеристики переходов и интенсивностей в ИК и КР спектрах. Выводятся правила отбора. Показывается, что сумма членов в формуле Гайзенберга–Крамерса может быть представлена в виде слагаемого, вычисление которого не приводится к суммированию, и быстро сходящегося ряда. Излагается квантовая теория электрооптических параметров, выясняется их физический смысл и тем самым строго обосновывается возможность расчётов колебательных спектров систем без ограничений на их размеры и структуру с помощью банков молекулярных фрагментов и валентно-оптической схемы. Эта схема расширяется на молекулярные ионы. Приводятся многочисленные следствия общей теории: правила отбора по симметрии, характеристичность интенсивностей, соотношения между интенсивностями в спектрах изотопозамещённых молекул, особенности интенсивностей полос поглощения обертонов, влияние на интенсивности полос в ИКС периферийных CH связей и скелета, зависимость интенсивностей полос и линий от числа одинаковых групп в молекулах и др.
Следующая глава содержит материал, связанный с общими, основанными на вариационной процедуре, методами решения ангармонических задач. Анализируется выбор потенциальной функции, влияние кинематической ангармоничности, применение гармонического, «уширенного» и морзевско-гармонического базисов и др. Обсуждается изотопический сдвиг при наличии ангармонизма. Излагается приближённый метод учёта ангармонизма для случая очень больших нерегулярных молекулярных образований.
Отдельное место в теории внутренних движений в атомах занимают внутренние вращения. Соответствующие вопросы рассматриваются в главе 15. В ней подробно исследуется проблема введения координат внутреннего вращения, отделение внутренних вращений от колебаний, интенсивности полос поглощения для вращательных переходов и др.
Последняя глава посвящена решению обратных задач. Обсуждаются различные варианты постановки таких задач, в частности, для спектров сложных молекул со слабо разрешенной структурой и многочисленными наложениями полос поглощения в ИК или линий в КР спектрах.
Книга будет опубликована в 2008 г.
б) Фрагментарный метод расчёта характеристик электронных состояний очень сложных молекул.
Характерной особенностью современной химии является быстро возрастающий интерес к исследованию и использованию для различных целей очень крупных молекул (молекулярных систем) с числом атомов до нескольких сотен. В таких объектах появляются свойства, не столь уж детально изученные на предшествующих этапах развития науки о микромире. Новая ситуация предъявляет и новые требования к теории, которая должна обеспечить возможность компьютерного анализа свойств молекулярных объектов практически без ограничений на их размеры.
Фундаментальное положение всей теории строения и свойств сложных молекулярных структур, которое является обобщением громадного эмпирического материала, состоит в том, что подавляющее число соединений можно «собрать» из отдельных крупных фрагментов в ранее изученных соединений. Это естественно приводит к проблеме создания таких методов расчётов характеристик крупных систем, которые изначально были бы ориентированны на использование результатов, накопленных в специальных банках данных. Такая проблема не была решена ранее.
Нами предложен простой метод формирования энергетической матрицы для задачи об электронных состояниях молекулярных систем любого размера, состоящих из отдельных крупных достаточно стабильных по своим характеристикам фрагментов. Показано, что соответствующие данные о фрагментах могут записываться в форме, позволяющей накапливать их в банках. В результате процесс определения отвечающих электронным движениям уровней энергии и собственных функций по своей логике становится аналогичным привычному приёму «сшивки» молекулы из фрагментов. При этом нет необходимости использовать чрезмерное число базисных АО.
в) Метод оценки вероятности структурных изомер-изомерных превращений при наличии большого числа квазивырождений уровней энергий взаимодействующих подсистем.
Ранее был обоснован подход к описанию молекулярных процессов, в частности, таких важных как процессы структурной изомеризации сложных систем, базирующийся на представлении о безызлучательных (безэнергетических) переходах между состояниями взаимопревращающихся подсистем (исходный и конечный структурные изомеры) при наличии очень близких по значениям электронно-колебательных уровней энергии взаимодействующих пар. В этом случае и при квантовом, и при классическом рассмотрении возникает колебательный процесс, имеющий близкую аналогию с классической передачей энергии от одного маятника к другому, если эти маятники связаны друг с другом и собственные частоты их колебаний одинаковы.
В 2007 г. показано, каким образом можно распространить развиваемую теорию структурных превращений молекул как результата резонансного смешивания электронно-колебательных волновых функций соответствующих состояний подсистем на случай произвольного числа резонирующих и квазирезонирующих уровней энергий.
г) Простая модель эффекта редупликации как следствие первых принципов.
Явление редупликации (репликации), заключающееся в точном самокопировании молекулярного объекта путём направленной организации и преобразования составляющих окружающей среды вызвало самое пристальное внимание после того, как было понято, что это явление определяет сам феномен жизни. Стадии самовоспроизведения достаточно хорошо изучены для важнейших случаев репликации ДНК и РНК. Представляет, тем не менее, интерес попытаться на основе первых принципов общей физической теории молекулярных превращений построить простую модель явления, позволяющую выявить не зависящие от конкретного молекулярного объекта условия, обеспечивающие процесс самовоспроизведения.
Это может дать ответ на принципиальные вопросы:
– Является ли явление репликации ДНК совершенно уникальным и возможным только для соответствующей системы или оно допустимо и в других случаях?
– Можно ли свести описания различных стадий всего процесса к простейшим и хотя бы количественно оценить вероятности каждого шага в общей последовательности с целью сформировать систему уравнений, позволяющую моделировать развитие явления во времени и производить компьютерные эксперименты?
Решению этих вопросов было посвящено специальное исследование.
На основании первых принципов предложена простая модель, описывающая процесс редупликации молекулярной структуры как последовательности стадий, вероятности которых могут быть оценены на основе общей теории молекулярных превращений. Показано, что передача генетической информации во времени (феномен жизни) в течение длительного периода невозможна без одновременного «эффекта смерти» и что учёт особенностей переходных состояний при реакциях (квантовые биения) приводит к появлению автоколебаний и биологического ритма.
д) Некоторые особенности проявления изотопного эффекта при структурных превращениях молекул.
Мономолекулярные межизомерные переходы молекул являются одной из весьма распространенных форм их структурных превращений. Хорошо известно, что при реакциях структурной изомеризации, как и других химических структурных превращениях молекул, изотопные эффекты во многих случаях играют весьма существенную роль и широко используются как при изучении хода реакции, так и при создании очень чувствительных аналитических методик. Простейшая теория изотопозамещения при химических реакциях (включая и реакции структурной изомеризации) основана на модели перевала через потенциальный барьер, разделяющий комбинирующие состояния (химические формы) молекулярных объектов. При этом игнорируется кинетическая стадия реакции. Это возможно для быстропротекающих реакций, где легко устанавливается термодинамическое равновесие. Однако так нельзя поступать при анализе реакций, идущих очень медленно, что характерно, например, для геохимических процессов.
Нами рассмотрен общий случай влияния изотопозамещения на характеристики структурных превращений молекул. Обращено внимание на вероятностный фактор, ранее не учитывавшийся в теории кинетического изотопного эффекта. Показано, что в ряде случаев он может оказаться определяющим и приводить к существенным, не только количественным, но и качественным, изменениям кинетики процесса структурного превращения: например, появлению или исчезновению эффекта индукции реакции и зависимости константы реакции от времени, инверсии соотношения количеств получаемых продуктов и др.
Влияние изотопозамещения на величины частот квантовых биений w, определяющих вероятности изомерных превращений молекул
|
Изомерные переходы
|
?A/?B
|
|
|
A
|
B
|
|
|
|
|
10 |
|
|
|
10-1 |
|
|
|
10-1 |
|
|
|
10-3 |
|
|
|
10-3 |
|
|
|
1
|
|
|
|
10-1 |
е) Поиск и исключение зависимостей между колебательными координатами при моделировании спектров больших молекул
Исследован алгоритм, позволяющий уверенно распознавать произвольное число сложных линейных зависимостей между колебательными координатами в молекулярной модели очень высокой размерности. Эти зависимости исключаются на этапе диагонализации кинематической части колебательного гамильтониана. Проведены компьютерные эксперименты, позволившие выработать оптимальные правила построения соответствующих вычислительных программ для работы с колебательным гамильтонианом очень высокой размерности.
ж) Простая модель описания ангармонических колебаний многоатомных молекул.
Проблема вычисления уровней энергии и вероятностей оптических переходов между ними для ИК спектров многоатомных молекул с ангармонической потенциальной функцией, допускающей диссоциацию системы, ранее была подробно рассмотрена в нашей исследовательской группе. Была предложена соответствующая аналитическая форма для потенциальной ямы с учётом разрыва концевых связей при их значительном удлинении, сформулирован и опробован на примерах конкретных молекул алгоритм решения ангармонической задачи прямым вариационным методом в морзевско-гармоническом базисе, созданы программы для ПК, позволяющие оперировать с моделями достаточно крупных (20-30 атомов) молекул. Сохраняется, однако, проблема учёта ангармонизма для систем, содержащих сотни атомов.
С этой целью предложены простая с точки зрения вычислений модель и метод расчёта уровней энергии и интенсивностей в ИК спектрах многоатомных молекул в ангармоническом приближении для достаточно высоких значений колебательных квантовых чисел, но с ограничением возможных движений ядер областью, ещё не достигающей выхода на диссоциационный предел. Подход не имеет ограничений на общий размер молекулярной структуры.Проблема вычисления уровней энергии и вероятностей оптических переходов между ними для ИК спектров многоатомных молекул с ангармонической потенциальной функцией, допускающей диссоциацию системы, ранее была подробно рассмотрена в нашей исследовательской группе. Была предложена соответствующая аналитическая форма для потенциальной ямы с учётом разрыва концевых связей при их значительном удлинении, сформулирован и опробован на примерах конкретных молекул алгоритм решения ангармонической задачи прямым вариационным методом в морзевско-гармоническом базисе, созданы программы для ПК, позволяющие оперировать с моделями достаточно крупных (20-30 атомов) молекул. Сохраняется, однако, проблема учёта ангармонизма для систем, содержащих сотни атомов.С этой целью предложены простая с точки зрения вычислений модель и метод расчёта уровней энергии и интенсивностей в ИК спектрах многоатомных молекул в ангармоническом приближении для достаточно высоких значений колебательных квантовых чисел, но с ограничением возможных движений ядер областью, ещё не достигающей выхода на диссоциационный предел. Подход не имеет ограничений на общий размер молекулярной структуры.
з) Разделение переменных и алгоритм поиска координат внутреннего вращения и кинетических коэффициентов в теории ядерных движений в многоатомных молекулах.
Внутренние вращения вокруг одиночных связей в многоатомных молекулах являются очень важным видом движений. Они, как известно, играют решающую роль в процессах согласования «ключ-замок» при биохимических реакциях с участием сложных структур. Этими же видами движений определяется и возможность некоторых изомер-изомерных превращений при сильном изменении геометрических форм исходных и конечных продуктов. В частности, это имеет место при превращении линейных углеводородов в разветвленные. Исследованию внутреннего вращения посвящено много монографий и статей. Казалось бы, не осталось никаких вопросов. Это, однако, не так. Как ни удивительно, до сих пор возникают проблемы с введением таких координат внутреннего вращения, которые приводили бы к максимальному разделению вращательных и колебательных движений.
Нами предложен универсальный алгоритм построения координат внутреннего вращения в многоатомных молекулах, основанный на свойствах матрицы кинематических коэффициентов при введении избыточной системы естественных координат. Рассмотрены приближения, при которых возможно разделение переменных. Указан точный вид оператора кинетической энергии при учёте внутренних вращений.
и) Матрицы смежности и графы химических превращений.
Громадное множество молекулярных объектов и химических превращений исходного продукта, оценить которые даже грубо на основании интуитивных соображений не представляется возможным, наводит на мысль о необходимости разработки простых приёмов своеобразного мониторинга реакций. Такая проблема впервые была поставлена и был обоснован возможный путь её решения.
Введены понятия матрицы смежности и графов химических превращений. В основу всех построений положена величина интеграла перекрывания волновых функций двух и более молекулярных структур. Разработанный алгоритм выполнения расчётов позволяет очень быстро рассматривать возможные пути взаимопревращений десятков и сотен объектов. Результаты обладают большой наглядностью и легко интерпретируются.
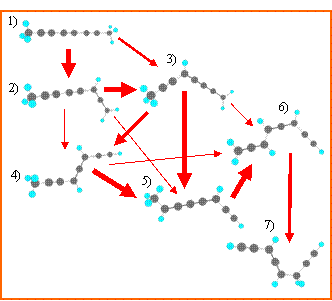
Матрица смежности и пути превращений
к) О некоторых закономерностях формирования органического вещества на ранних стадиях геохимической эволюции.
Проблема возникновения на Земле сложного органического мира, вплоть до появления тех процессов, которые определяют саму возможность феномена жизни, является одной из интереснейших в общем направлении познания окружающего мира. Общеизвестными фактами является то, что, во-первых, все жизненные процессы совершаются в молекулярном мире и, во-вторых, этот молекулярный мир создавался в результате постепенного усложнения его составляющих (молекулярных структур), начиная от простейших. Вряд ли можно проследить в деталях всю эволюцию образования условий, приводящих к возникновению в молекулярном мире таких процессов, которые трактуются как жизненные, однако можно надеяться, что некоторые важнейшие закономерности можно уловить, если исходить из так называемых первых принципов.
Впервые показано, что самая ранняя стадия образования более сложных, чем исходные, органических молекул подчиняется закону возрастания энтропии в замкнутой системе. Этот результат принципиально отличен от общепринятого представления, заключающегося в утверждении, что любая самоорганизация возможна только в открытых системах. Причина заключается в том, что при образовании из простейших молекул более сложных образуются химические связи между углеродными атомами, что приводит к появлению низкочастотных колебаний и резкому возрастанию колебательной составляющей общей энтропии ансамбля. С ростом размеров структур этот эффект убывает. Поэтому дальнейшая самоорганизация становится возможной только в открытых системах.
л) Метод оценки влияния внешнего электрического поля на вероятности фотохимических превращений.
Хорошо известен эффект влияния сильного электрического поля на ход (выход продукта) фотохимических реакций. Этот эффект проявляется как при фотохимических реакциях при большой плотности квантов инициирующего облучения, так и при наложении статического поля со значительной напряжённостью.
Предложен подход к оценке действия сильного внешнего электрического поля на вероятности фотохимических превращений. Подход основан на анализе возникновения или исчезновения необходимого для химического превращения резонанса уровней взаимодействующих подсистем и изменения интеграла перекрывания соответствующих волновых функций.
м) Спектроскопическое вычисление энергий диссоциации связей CH и OH.
С помощью спектроскопического и квантово-химического методов определены энергии диссоциации связей СН и OH в альдегидах, кетонах, кислотах, спиртах, фторпроизводных метана, этана, этена, пропена и бензола. Спектроскопические значения энергии диссоциации связей CH и OH рассчитывались на основе фундаментальных полос поглощения в ангармоническом приближении вариационным методом с использованием морзевско-ангармонического базиса. Квантово-химические вычисления производились с использованием 6-311G(3df,3pd)/B3LYP базиса. Получены закономерности изменения значений энергии диссоциации связи при изменении структуры молекулы и проведено их детальное обсуждение.
н) Влияние внешнего температурного воздействия на ход химической реакции.
Показано, что путем импульсных тепловых воздействий, изменяющих температуру ансамбля молекул в процессе реакции, можно целенаправленно управлять ходом медленной (с характерными временами структурных превращений порядка минут и более) термической реакции в молекулярных ансамблях, изменяя тем самым соотношение количеств ее продуктов и само направление процесса в случае разветвляющихся цепочек превращений.
При определенных условиях практически стационарное, но инверсное по сравнению с равновесным больцмановским, распределение молекул по уровням энергии может поддерживаться в молекулярном ансамбле в течение очень длительного времени, на много порядков величин превышающем время тепловой релаксации. Для сложных разветвляющихся цепочек химических превращений возможны «переключения» направления хода процесса с одной на другую ветвь при определенных температурных режимах. Температурные режимы могут варьироваться в широких пределах от простейшего в виде прямоугольного импульса до сложных периодических. Выбор наиболее оптимального в каждом конкретном случае (форма и количественные характеристики) может осуществляться методами молекулярного моделирования.
Изучен вопрос о влиянии внешнего периодического температурного воздействия на выход продукта химической реакции. Показано, что при определённом соотношении частоты резонансных переходов между реагирующими подсистемами выход продукта может увеличиться на 10-15%.
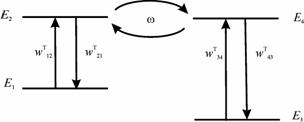
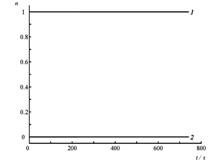
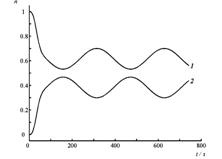
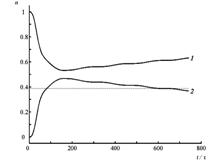
Четырехуровневая двухизомерная молекулярная система.
Состояния E1 и E2 отвечают изомеру 1, E3 и E4 – изомеру 2.
|
(а)
 |
(б)
 |
|
(в)
 |
(г)
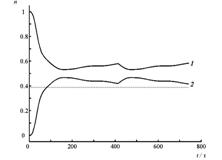 |
Кинетические кривые для суммарных заселенностей состояний изомеров 1 (1) и 2 (2) четырехуровневой модели температурной реакции изомеризации при начальном заселении изомера 2. Температура равна ?T1=0.2?E21 /kB(a), T2=10T1 (б), изменяется во времени «импульсно» (T=T1 при t<0, t>160 ?; T=T2 при 0?/2?320? на временных отрезках ?t ? T?/2 ? 8 ? 80?. Штриховой линией показано среднее значение, отвечающее стационарному температурному режиму T=T2
Указано, что при медленных температурных реакциях в сложных молекулах эффекты безызлучательных переходов вследствие квантовых биений резонансных состояний комбинирующих молекулярных структур можно наблюдать в обычных (без высокого временного разрешения) ИК спектрах поглощения в виде осцилляций интенсивностей отдельных линий. Предложен способ обеспечения необходимого уровня когерентности осцилляций в ансамбле молекул, опирающийся на использование импульсного нагрева молекулярной системы. Регистрация качественно новых эффектов методами традиционной ИК спектроскопии, включая и их количественные характеристики, существенно повышает информативность получаемых спектральных данных и открывает возможность применения таких высокоразвитых экспериментальных методов для изучения химических реакций, включая и их динамику. Круг этих реакций весьма широк, охватывает как «лабораторные», так и «природные» (геохимия, экология и др.) процессы с характерными временами порядка минут и более.
Периодические внешние воздействия на способную к биохимическим преобразованиям молекулярную систему приводят к возникновению эффекта параметрического резонанса и, как следствие, при определенных условиях к получению выигрыша в количестве продуктов реакции и к характерным частотным и фазовым зависимостям. Вследствие этого в ансамбле молекул может осуществляться естественным путем синхронизация внутримолекулярных колебательных процессов и возникать общий для всего ансамбля биологический ритм, причем адекватный периодическим изменениям внешних условий и «управляемый» ими. Можно полагать, что периодические изменения характеристик внешних воздействий выступают как стимулирующие биохимические процессы и, более того, как факторы, способные не только влиять на «направление эволюции» биомолекулярной системы, но и определять его.
о) Моделирование вибронных спектров сложных молекул
В рамках второго приближения параметрического метода проведен расчет структуры молекул пиридина и дипиридилэтилена в возбужденном состоянии и их электронно-колебательных спектров. Полученная система параметров, включающая в себя параметры сигма- и пи-типа, обеспечивает количественное согласие теоретических спектров пиридина и дипиридилэтилена с экспериментальными, что свидетельствует об адекватности моделей реальной структуре молекул. Параметризация носит достаточно полный характер и позволяет моделировать на количественном уровне колебательную структуру спектров сложных молекул, содержащих аналогичные фрагменты, как для сигма-пи*-, так и пи-пи*-переходов. Показана высокая устойчивость системы параметров аценовых и полиеновых фрагментов при столь существенных замещениях атомов в молекулах. Проведена интерпретация колебательной структуры электронных спектров рассмотренных молекул и проанализированы изменения их структуры при возбуждении.
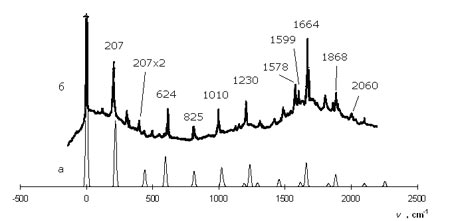
Рассчитанный (а) и экспериментальный (б) спектры флуоресценции дипиридилэтилена
См. также: Результаты 2007 г