
Общая теория молекулярных процессов
1) Предложены методы решения задачи моделирования реакции разложения сложной молекулы и поиска колебательных волновых функций, необходимых для вычисления вероятности прохождения этой реакции в различных физических условиях.
Показано, что для вычисления вероятности реакции разложения можно пользоваться разработанными ранее приемами моделирования реакций изомерных превращений. С помощью таких приемов находятся матрицы соотношения между нормальными координатами исходной и предраспадной молекулярной модели. Это соотношение используется для вычисления интеграла перекрывания колебательных волновых функций двух моделей. При нормировке волновых функций используются данные о температуре и плотности среды, в которой происходит реакция разложения.
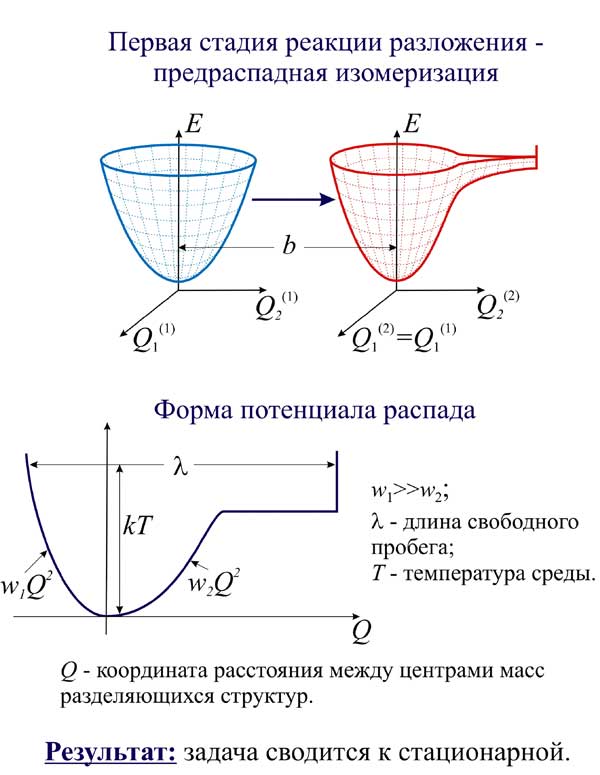
2) Поставлены и решены задачи моделирования и предсказания качественных и количественных особенностей кинетики процессов молекулярных превращений и их спектральных проявлений в широких спектральном и временном диапазонах (включая и фемтосекундные фотохимические процессы).
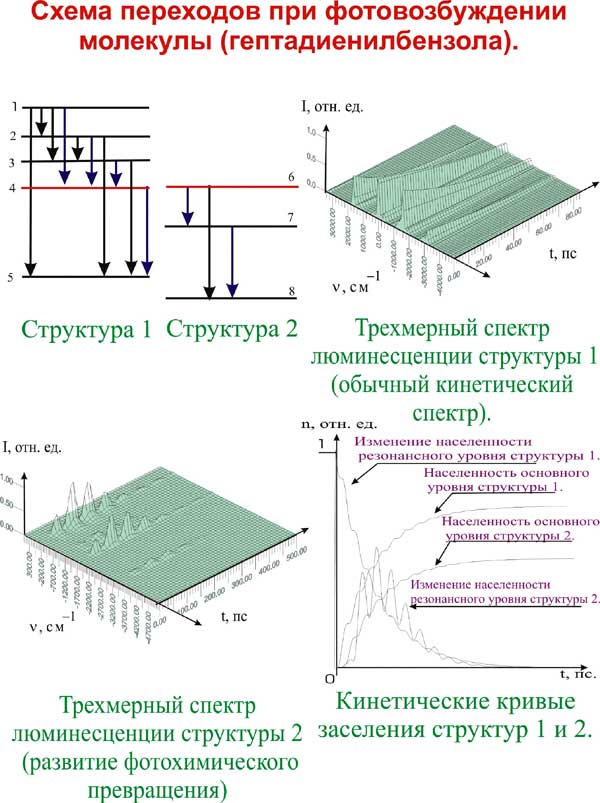
а) Показано, что кинетика внутримолекулярных процессов и спектров с временным разрешением с учетом квантовых биений резонирующих состояний изомеров или выделенных подсистем уровней одной изомерной формы может быть описана с использованием молекулярной модели, интерпретирующей эффект биений как безызлучательный переход.
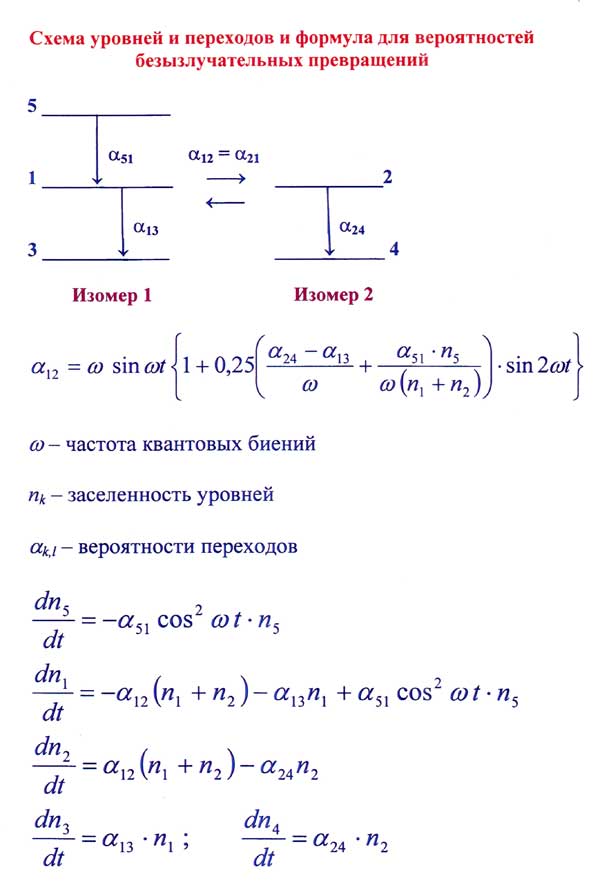
Получено выражение для вероятности безызлучательного перехода, которая прямо пропорциональна частоте биений и зависит от времени осцилляторно, моделируя таким образом временную зависимость процесса перехода от одной подсистемы к другой. Параметром модели молекулярной системы является определяющая вероятность безызлучательного перехода частота биений, непосредственно связанная с параметром, характеризующим внутримолекулярные межизомерные взаимодействия (соответствующий недиагональный элемент энергетической матрицы).
Характер изменения заселенностей уровней и, соответственно, интенсивностей полос в спектрах в предлагаемой модели хорошо согласуется с наблюдаемым в эксперименте, включая и тонкую структуру временных зависимостей – осцилляции интенсивностей линий. При анализе временного эксперимента с высоким разрешением необходим учет аппаратной функции, приводящей к количественным и качественным изменениям временных зависимостей.
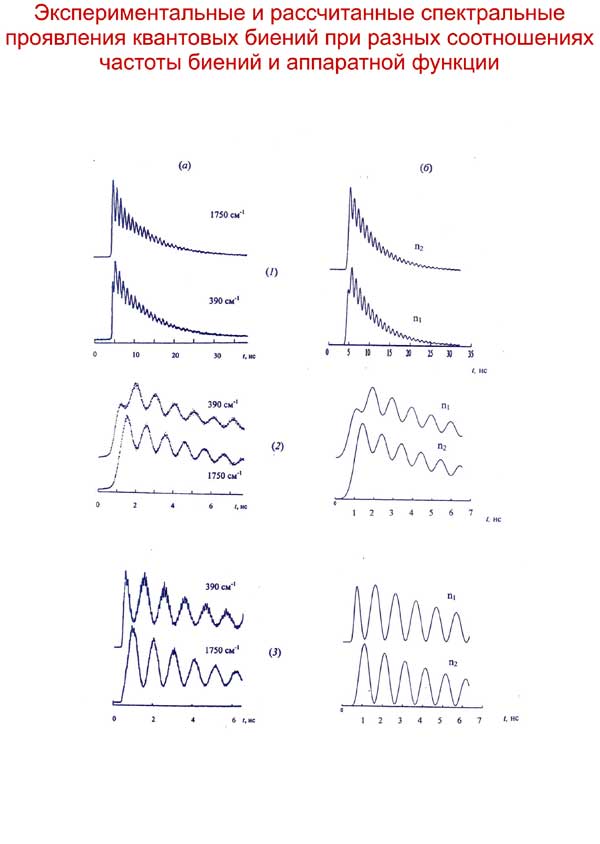
Показано, что традиционная модель безызлучательных переходов с постоянной по величине вероятностью имеет существенно ограниченную область применения – очень высокие по сравнению с вероятностями оптических переходов частоты биений.
б) Разработан метод вычисления интегралов наложения колебательных волновых функций комбинирующих состояний изомеров, определяющих частоты квантовых биений и вероятности межизомерных переходов. Развитые методы обеспечивают проведение достаточно быстрого и массового моделирования процессов химических превращений и создают возможность выполнения компьютерных экспериментов предсказательного характера. Показана также возможность проведения грубых оценок вероятности межизомерного перехода (да/нет) для сложной молекулярной системы.
в) На основе разработанного ранее метода расчета динамических электронно-колебательных спектров сложных молекул с учетом изомерных преобразований развит более общий вариант, обеспечивающий возможность моделирования спектров нескольких изомерных форм, число которых может быть любым в зависимости от поставленной задачи. Возможен анализ динамики населенности уровней энергии изомеров. Разработаны и оптимизированы вычислительные алгоритмы, а также создано специализированное программное обеспечение для ПК и суперкомпьютера (типа МВС 1000). Проведены компьютерные эксперименты и показана возможность моделирования в масштабе реального времени динамических спектров и кинетических внутримолекулярных процессов для сложных соединений с учетом их межизомерных преобразований.

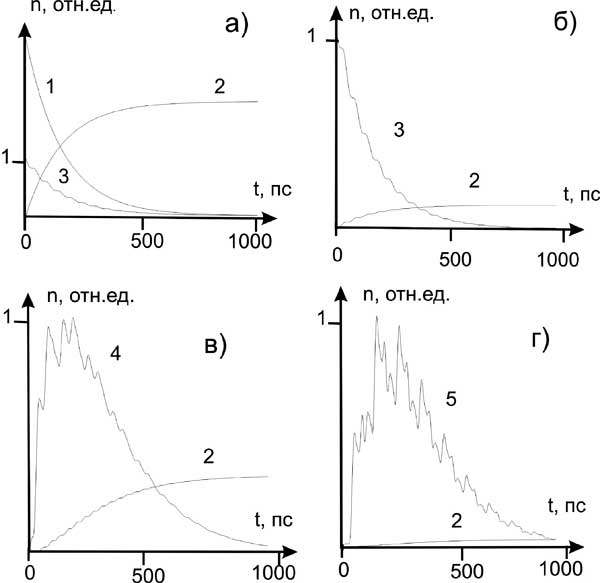
г) Проведены компьютерные эксперименты для моделей реальных молекулярных структур, содержащих несколько десятков атомов, при которых решались системы кинетических уравнений размерностью порядка 1000 с учетом более 30000 оптических переходов, влияющих на динамику молекулярной системы.
Обнаружено, что ход фотохимического процесса существенно зависит от величин молекулярных параметров и может качественно меняться в относительно малой области вариации их значений. Это коррелирует с экспериментальными данными о высокой чувствительности таких процессов к небольшим изменениям молекулярных структур или внешним воздействиям.
3) Разработана молекулярная модель для описания кинетики процессов внутримолекулярных преобразований и их спектральных проявлений при термическом возбуждении.
Показано, что данная модель адекватно отражает (и предсказывает) наблюдаемые на опыте зависимости, а именно: равновесное распределение молекул по уровням после протекания реакции; количество молекул в разных изомерных формах; температурную зависимость констант скорости реакции термической изомеризации в очень широком диапазоне температур в соответствии с законом Аррениуса.
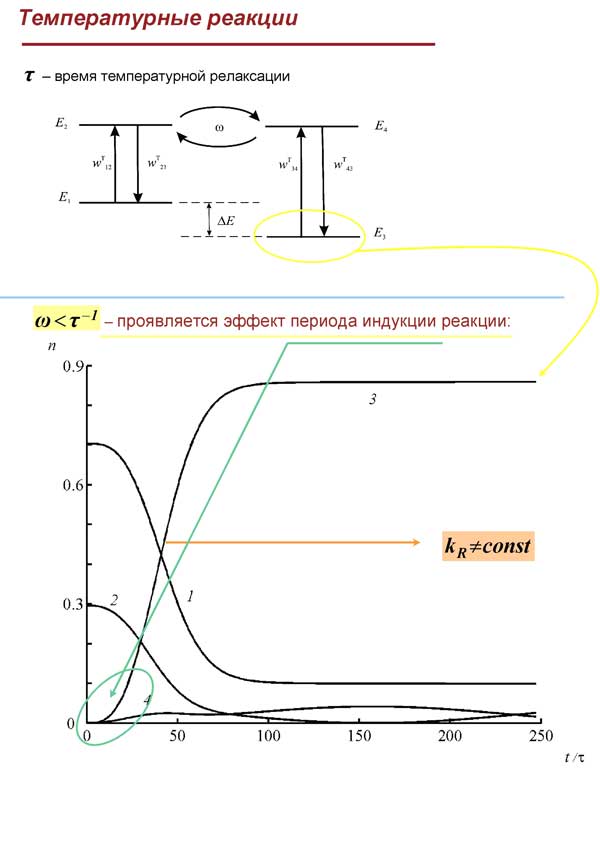
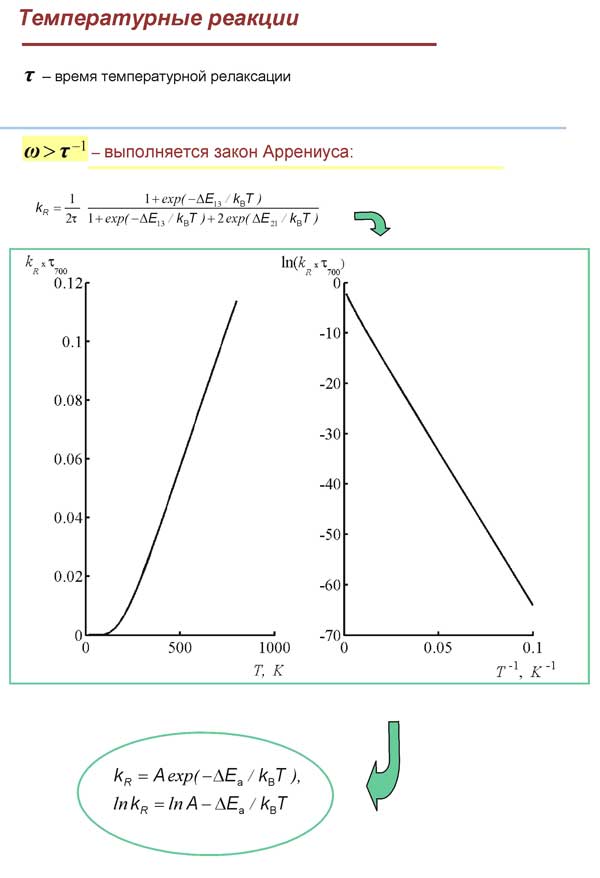
Показано, что в отличие от типичных в химии быстрых реакций в кинетике медленных реакций явно проявляются осциллирующие факторы, поэтому при соответствующей постановке спектрального эксперимента величины частот биений и значения матричных элементов внутримолекулярных “межизомерных” взаимодействий могут быть определены опытным путем.
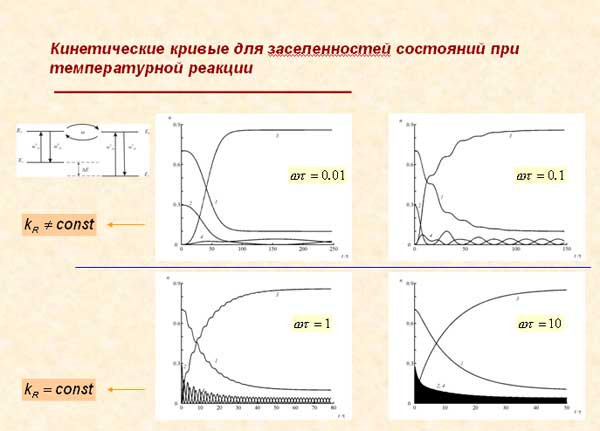
4) Выявлены качественные и количественные особенности влияния на кинетику фотохимических и тепловых реакций и на их спектральные проявления структурных молекулярных изменений и изотопозамещений.
Показано, что в теории кинетического изотопного эффекта необходимо учитывать не только влияющий на него энергетический фактор (положения уровней энергии и их изменения при изотопозамещении), но и вероятностный, обусловленный изменениями вероятностей безызлучательных переходов при изотопозамещении из-за изменений форм нормальных колебаний. Второй фактор может приводить к весьма значительным по величине эффектам и оказывается в ряде случаев решающим, приводящим к изменениям не только количественных характеристик эффекта, но и качественных, вплоть до изменения его знака, поскольку вероятность переходов может изменяться на порядки величин.
5) Показано, что наблюдающиеся низкочастотные периодические процессы в спектроскопии и химических превращениях, протекающие без изменения полной энергии сложной молекулярной системы, могут быть объяснены как результат синхронизированных периодических безызлучательных переходов между резонирующими основными состояниями квантовых систем разной структуры.
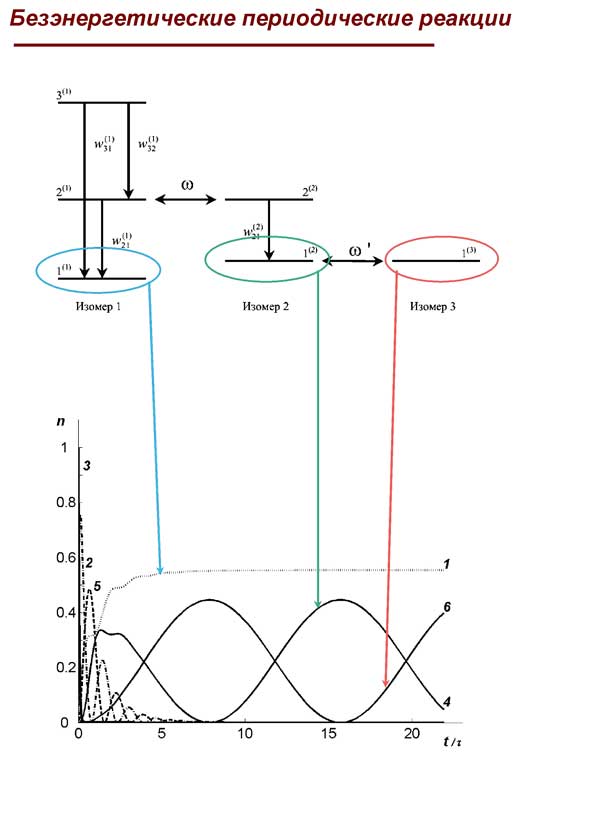
6) Изучены особенности и количественные характеристики вероятностей структурных преобразований сложных молекул, связанные с локализацией колебаний в малых частях молекулярного пространства. Выяснена роль внутримолекулярного окружения реакционных центров на ход локализованных фотохимических превращений молекул.
а) На основе базовых положений квантовой теории без привлечения эмпирических данных, т.е. из первых принципов, уточнено понятие реакционного центра, проведен анализ необходимых и достаточных условий его появления и специфики протекающих в нем процессов. Показано, что (учитывая связь величины структурного преобразования молекул с вероятностью такого превращения) появляются «пространственные» запреты на ход как фотохимических реакций изомеризации, так и реакций других типов, и что локальность структурных преобразований, затрагивающих, в основном, относительно небольшую атомную группировку молекулы (ее фрагмент), является одним из ключевых факторов, определяющих возможность данного межизомерного перехода.
Очень значительные структурно-изомерные преобразования молекул возможны только в результате ряда независимых последовательных шагов, каждому из которых отвечают локальные деформации. Этим же объясняется невозможность прямого превращения A+B?C+D, а допустимо лишь превращение A+B?F?F*?C+D или через серию последовательных изомерных преобразований.
б) Показано, что реакционные центры в хорошем приближении действуют независимо от молекулярного окружения. Изменения вероятностных характеристик, определяющих процесс реакции, быстро затухают в ряду молекул (например, при увеличении их размеров) и, таким образом, локальные свойства реакционного центра существенно не изменяются.

в) Ход химического процесса и свойства реакционного центра могут измениться, если молекула способна к не требующим особых энергетических затрат изменениям пространственных характеристик, подготовляющих собственно химический процесс (например, внутренних вращений). Этим, например, объясняется возможность структурного преобразования углеводородов от линейных к Т-образным формам.
г) Анализ и модельные расчеты показали, что важнейшую роль для возможности (т.е. относительно высокой вероятности) структурного превращения молекул играет не только свойство его локальности, но и наличие отвечающих такому структурному преобразованию характеристических колебаний. Количественными показателями наличия таких колебаний служат легко вычисляемые векторы сдвига, описывающие структурные деформации молекулы при химических превращениях в системе нормальных координат. Чрезвычайно важно, что особенности движений атомов при характеристических колебаниях четко проявляются в колебательных и электронно-колебательных спектрах в виде полос поглощения, сохраняющих свои положения на шкале волновых чисел, а часто и свои интенсивности.
Тем самым указан путь поиска реакционных центров многоатомных молекул и выявления их спектральных признаков, что принципиально важно при планировании эксперимента. Разработанные в рамках данного цикла исследований методы расчета и компьютерные программы обеспечивают возможность проведения таких прогностических вычислительных экспериментов.
7) Известно, что интерпретация экспериментально наблюдаемых фотохимических спектров составляет для многоатомных систем крайне сложную и в прошлом часто неразрешимую задачу. Это вообще является важнейшим ограничивающим фактором развития фотохимии как метода исследования молекулярных превращений и управления процессом этих превращений путем дополнительных оптических воздействий. Решение этих проблем возможно только на базе детального теоретического рассмотрения и широкого применения компьютерного эксперимента. Для интерпретации спектров обычной флуоресценции и поглощения успешно применяется развитый параметрический метод, так что получаемые теоретические электронно-колебательные спектры хорошо количественно согласуются с экспериментальными.
Выяснена степень применимости разработанных ранее методов моделирования возбужденных состояний молекул для прогнозирования спектрального эксперимента с резонансным возбуждением, типичного в фотохимии.
Показано (на примере молекулы антрацена, для которой имеются весьма полные данные о дисперсных спектрах), что среди наблюдаемых спектров флуоресценции можно выделить по типичным свойствам колебательной структуры и степени ее сложности три характерные группы.
К первой группе относятся спектры флуоресценции из вибронных состояний, переход в которые из основного или маловероятен (слабые по интенсивности полосы спектра поглощения), или запрещен по симметрии. Такие спектры имеют вибронную структуру, повторяющую по частотному составу и распределению интенсивности спектр обычной флуоресценции.
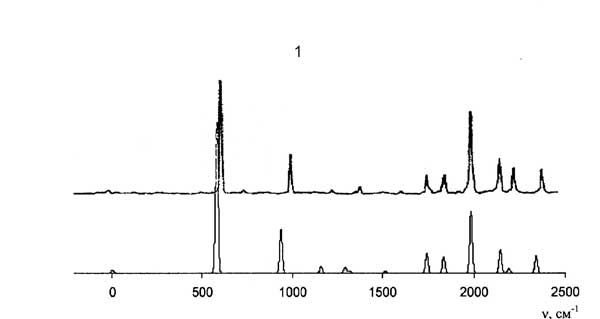
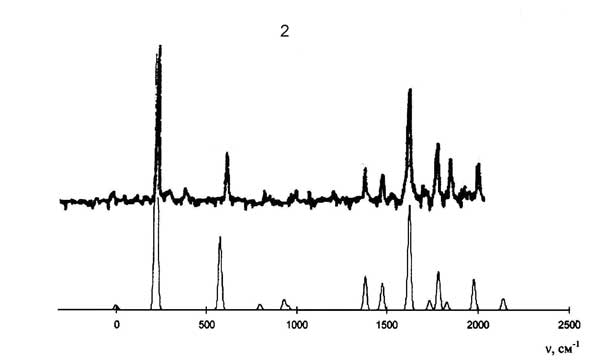
Экспериментальные (сверху) и рассчитанные (снизу) спектры дисперсной флуоресценции антрацена при возбуждении состояний S1+583 см-1 (1) и S1+209 см-1 (2).
Вторую группу составляют спектры, возбуждаемые в вибронные состояния, переход в которые разрешен и имеет большую интенсивность. Их структура более сложная и содержит несколько групп спектральных полос, наложенных друг на друга и начинающихся от сравнимых по интенсивности обертонов возбуждаемого колебания.
В третьей группе объединяются спектры, не относящиеся к первым двум, поскольку их структура содержит дополнительные полосы, требующие специального анализа.
Путем прямого расчета можно провести полную и детальную интерпретацию экспериментальных спектров первого и второго типа. Для спектров третьего типа, интерпретация которых крайне затруднена, компьютерное моделирование позволяет получить наиболее вероятное описание колебательной структуры.
Теоретические спектры количественно согласуются с экспериментальными, что свидетельствует об эффективности метода при их моделировании. Расчеты данным методом носят полностью предсказательный характер, так как параметризация молекулярных моделей получена с использованием экспериментальных спектральных данных иного типа.
См. также: Результаты 2003-2006 гг